Researchers Conduct the First Meta-Analysis of Cell-Lineage Specific DNA Methylation Changes in Blood
Smoking is a major risk factor for many complex diseases, including various cancers and cardiovascular disease. Epigenetic changes, and DNA methylation alterations in particular, are believed to play an important role, potentially mediating the risk of disease.
In order to understand how smoking affects DNA methylation (DNAm) in normal cells, many previous Epigenome-Wide Association Studies (EWAS) have mapped such DNAm alterations in whole blood, an easily accessible tissue. However, because whole blood is a heterogeneous tissue, composed of many different cell-types, interpretation of DNAm changes is hampered, as it is unknown which particular cell-types may be affected.
A team led by Professors AndrewTESCHENDORFF and WANG Sijia, both from the CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health of the Chinese Academy of Sciences, have now addressed this problem, by performing the first ever meta-analysis of whole blood EWAS in a cell-lineage specific manner. The team focused on smoking and assembled DNAm data from 7 independent EWAS, including the first Chinese cohort where smoking associated DNAm changes have been comprehensively mapped.
The analysis of the data revealed that smoking-associated DNAm changes are largely independent of ethnicity, justifying the merging of Chinese and white Caucasian cohorts for a meta-analysis. The meta-analysis further revealed how most of the DNAm changes in blood occur within the myeloid lineage, mapping preferentially to DNase hypersensitive sites (DHS) characteristic of inflammatory monocytes and macrophages. Importantly, the analysis also revealed a novel myeloid-specific DNAm signature associated with acute myeloid leukemia, a hematological cancer for which smoking is a moderate risk factor. In contrast, the study did not find many DNAm alterations within the lymphoid lineage.
According to Andrew TESCHENDORFF, this study is important because “Our study demonstrates that it is possible to use a computational approach to infer cell-type specific DNAm changes, thus avoiding the need for extremely laborious and expensive cell-sorting”. He further adds that “We hope that our study may serve as a paradigm for performing similar meta-analysis of EWAS in a cell-type specific manner”.
WANG Sijia, who co-led the investigation, states “This study is highly significant because the computational strategy is flexible and easily adaptable to other risk factors and exposures such as obesity, air pollution and alcohol consumption, and importantly also adaptable to other tissues like skin or buccal swabs”.
This work was published in Nature Communications under the title “A cell-type deconvolution meta-analysis of whole blood EWAS reveals lineage-specific smoking associated DNA methylation changes” on 22nd Sept. 2020.
This work is funded by the Chinese Academy of Sciences, National Natural Science Foundation of China, the Shanghai Municipal Science and Technology Major Project, Ministry of Science and Technology, National Key Research and Development Project, etc.
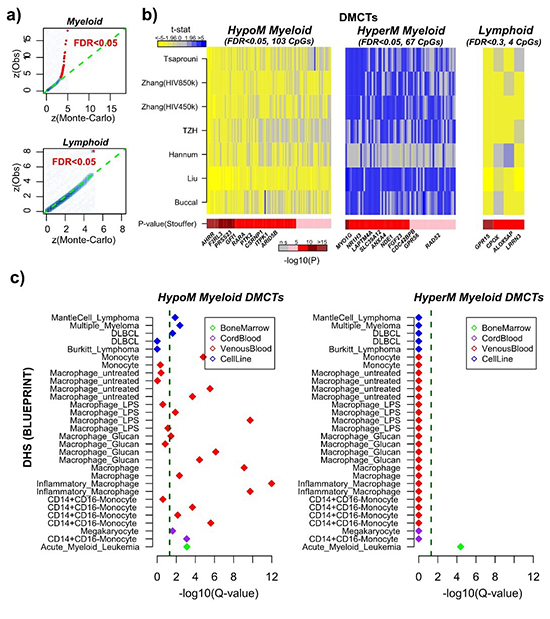
Meta-analysis reveals how most of the smoking-associated DNA methylation changes in blood occur within the myeloid lineage. (Image by Professors Andrew TESCHENDORFF and WANG Sijia's team)
Media Contact:
WANG Jin (Ms.)
Shanghai Institute of Nutrition and Health,
Chinese Academy of Sciences
Email: sibssc@sibs.ac.cn