Researchers Enable Computational Single-cell Dissection of Epigenomes for Arbitrary Tissue-types Without Need for Experimental Cell-sorting
The epigenome is altered in a wide range of diseases and in response to disease risk factors. A particular type of epigenetic alteration known as DNA methylation (DNAm), representing a covalent modification of DNA, is believed to be causally implicated and holds great promise as a disease biomarker. Due to cost and experimental limitations, measuring DNAm in large numbers of samples has only been possible at the bulk-tissue level, which averages out the DNAm changes across the many underlying cell-types, thus hampering power and obscuring biological interpretation. There is therefore an urgent need to develop cost-effective computational methods that allow dissection of bulk-tissue DNA methylomes at single-cell resolution.
Prof. Andrew TESCHENDORFF and Ms ZHU Tianyu, a PhD student in Prof. Teschendorff's lab at the CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health of the Chinese Academy of Sciences, have now cracked this outstanding computational problem, developing a novel statistical algorithm called “EpiSCORE”, which allows the precise cellular composition of arbitrary tissue-types to be inferred, as well as identifying the specific cell-types where disease-associated DNAm alterations occur.
Two fundamental and original ideas underlie the development and success of EpiSCORE. First, it leverages the power and high-resolution nature of existing and upcoming tissue-specific single-cell RNA-Seq atlases, as generated by the Human Cell Atlas Consortium. Second, it translates a tissue-specific mRNA expression atlas into a corresponding tissue-specific DNAm atlas, at effectively single-cell type resolution. The researchers demonstrate how this translation of information from mRNA expression to DNA methylation is possible, precisely because many of the cell-type specific markers exhibit underlying differences in DNA methylation. They applied EpiSCORE to lung cancer to reveal novel epigenetic alterations in lung cancer endothelial cells, suggestive of an endothelial-to-mesenchymal transition, underpinning invasion and metastasis.
What makes this study particularly significant is the fact that the great majority of DNAm data residing in public repositories has been generated at the bulk-tissue level, which means that without tools like EpiSCORE, the potential of this great data resource remains unrealized. EpiSCORE now enables other researchers to re-analyse these datasets allowing the precise cellular compositions of these tissues to be determined, as well as the identification of cell-type specific DNAm changes, an important task for elucidating the causal role of epigenetic changes in disease aetiology.
Moreover, EpiSCORE presents a cheap, cost-effective alternative to performing single-cell methylomics. Thus, unlike single-cell methylomics, EpiSCORE is scalable to large EWAS performed in arbitrary tissue-types.
This work was published in Genome Biology under the title “EPISCORE: cell-type deconvolution of bulk tissue DNA methylomes from single-cell RNA-Seq data” on 4th of September, 2020. It was funded by the Chinese Academy of Sciences and National Natural Science Foundation of China.
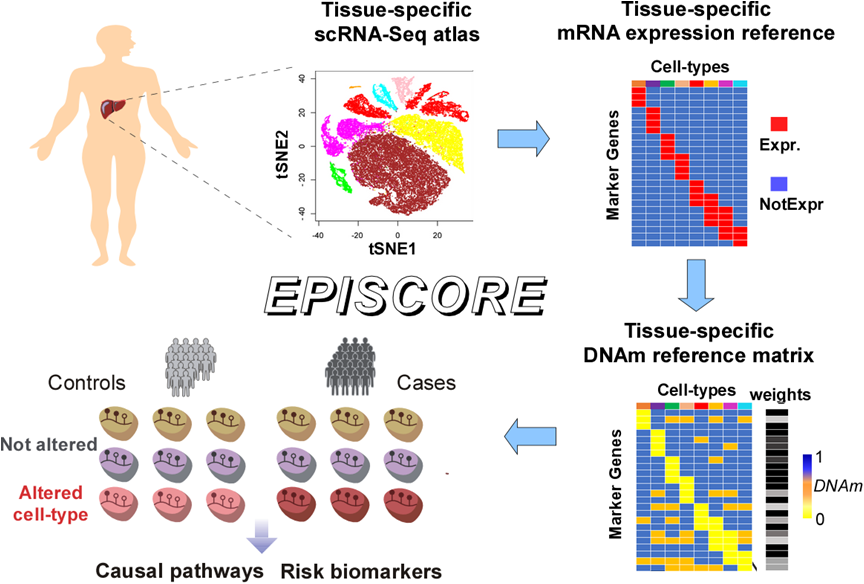
EPISCORE Workflow (Image by Prof. Teschendorff's team)
Media Contact:
WANG Jin (Ms.)
Shanghai Institute of Nutrition and Health,
Chinese Academy of Sciences
Email: sibssc@sibs.ac.cn